



peuvent devenir visibles chez l'être humain.


hypertropie (déviation vers le haut), hypotropie (déviation vers le bas)



L’Incontinentia Pigmenti (IP), aussi appelé syndrome de Bloch-Sulzberger, est une génodermatose rare (maladie de peau d’origine génétique), qui se manifeste par des atteintes au niveau de la peau, des dents, des yeux et du système nerveux central.
L’IP fait partie de la famille des dysplasies ectodermiques caractérisée par une absence d’au moins 6 dents, des atteintes sur la forme des dents, une malformation cranio-faciale et d’autres anomalies des tissus dérivés de l’ectoderme.
Le nom de la maladie provient de l’absence ou d’anomalies pigmentaires observées sur la peau à l’examen microscopique (incontinence pigmentaire).
Historiquement, l’Incontinentia Pigmenti a été étudié par le dermatologue suisse Bruno Bloch (à gauche) et américain Marion Sulzberger (à droite) en 1926, le dermatologue allemand Hermann Werner Siemens en 1929, l’ophtalmologiste suisse Adolphe Franceschi et le dermatologue allemand Josef Jadassohn en 1954.
L’Incontinentia Pigmenti est une maladie génétique liée à une mutation du gène NEMO (NF-kappa-B essential modulator) situé sur le chromosome X.
Dans 10 à 25% des cas, la transmission de la mutation se fait par les parents, selon « le mode dominant lié à l’X », mais peut aussi, dans 50% des cas, survenir de manière sporadique, sans antécédents familiaux de la maladie, on parle alors de mutation sporadique dite de « novo ».
Comme la mutation se situe sur le chromosome X, les garçons porteurs de la mutation souffrent de symptômes très sévères de la maladie, qui est généralement létale in utéro. Les filles, en revanche, présentent une gamme plus large de symptômes, d’expressivité variable allant d’une légère pigmentation cutanée à des problèmes dentaires et oculaires.
Un diagnostic prénatal permet de déceler l’Incontinentia Pigmenti mais pas d’en prévoir la gravité. La prévalence à la naissance de cette maladie est d’environ 1 sur 143 000 naissances.
Les symptômes de l’Incontinentia Pigmenti varient considérablement d’une personne à l’autre, mais voici les principales manifestations de la maladie.
Les manifestations cutanées
Les manifestations cutanées le long des lignes de Blaschko constituent un critère majeur du diagnostic clinique. Elles sont classées en 4 stades successifs dans le temps, certains stades peuvent être absents ou coexister chez le patient.
Stade 1, vésiculaire : de la naissance à l’âge de 4 mois, ce stade se manifeste par le développement de cloques ou de vésicules sur la peau, qui sont souvent douloureuses et peuvent entraîner des infections cutanées.
Stade 2, verruqueux : entre 2 et 6 mois, ce stade se caractérise par l’apparition d’excroissances ressemblant à des verrues le long des lignes de Blaschko. Certaines de ces lésions persistent à l’âge adulte.
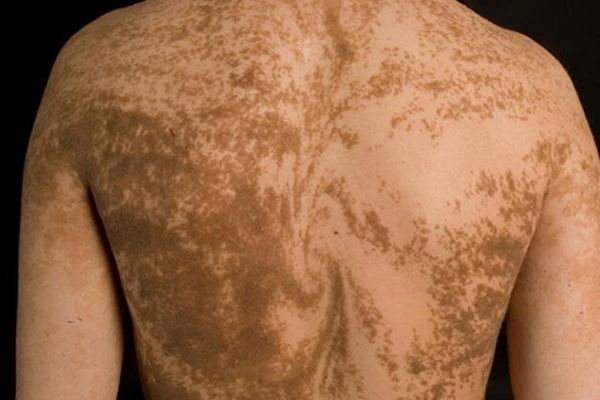
Stade 3, hyperpigmentation : débute après 6 mois, parfois après l’âge de 2/3 ans, ce stade « pigmenté », le plus spécifique donne son nom à la maladie et entraîne une hyperpigmentation visible « en tourbillon, jet d’eau, ou confettis ». Ces lésions spécifiques disparaissent en général complètement après la puberté.
Stade 4, hypopigmentation : débute à l’âge adulte mais peut exister dès les premiers mois de la vie en laissant apparaître des stries ou des taches hypopigmentées ainsi qu’une alopécie. L’hypopigmentation correspond à la diminution ou l’absence de synthèse de la mélanine par les mélanocytes (cellules de l’épiderme).
Aux manifestations cutanées, peuvent aussi s’ajouter des atteintes au niveau du cuir chevelu, des ongles et parfois des glandes mammaires (10% des cas).
Les atteintes dentaires
Les atteintes dentaires sont les plus fréquentes manifestations extra-cutanées de l’IP, tant au niveau du nombre que de la forme des dents.
L’anodontie (absence de dents) partielle ou totale sur les incisives ou les pré-molaires est très fréquente (90% des cas), on retrouve aussi des anomalies de nombre (dents surnuméraires), des anomalies de formes (dents coniques), ainsi qu’une altération de l’e-mail. Ces atteintes dentaires peuvent concerner la dentition temporaire et/ou définitive.
Enfin, il a été rapporté des fentes du palais (labiale et/ou palatine).
Les atteintes oculaires
Les atteintes oculaires représentent 30% des cas et débutent en général dès la 1ère année de vie, avec des anomalies comme un strabisme (ésotropie, exotropie, hypertropie, hypotropie), une atteinte du globe oculaires ou du nerf optique, ainsi qu’une cataracte.
Les atteintes du système nerveux central représentent 20 à 30% des cas, avec des manifestations comme un retard mental, un retard moteur, une insuffisance du développement du cerveau, une survenue de crise d’épilepsie ou de convulsions, et très rarement des anomalies de développement des os.
Le diagnostic de l’Incontinentia Pigmenti repose sur l’examen clinique notamment dermatologique et dentaire.
L’apparition précoce de vésicules et de pustules comparables à celle de la varicelle, sur la peau du nouveau-né suffit à poser le de diagnostic. Les anomalies dentaires sont aussi considérées comme un critère majeur, confirmées par un bilan oral et radiologique précoce.
Le diagnostic est ensuite confirmé par une analyse génétique. Pour les femmes atteintes d’IP, un Diagnostic PréNatal (DPN) est fortement conseillé.
La prise en charge de l’Incontinentia Pigmenti repose d’abord sur un suivi dermatologique en concertation avec une équipe pluridisciplinaire (chirurgien dentiste, ophtalmologiste, neurologue…) et une équipe paramédicale (kinésithérapeute, orthophoniste, ergothérapeute…). Le suivi étant bien sûr en fonction des atteintes, différent d’un patient à l’autre.
Au niveau des atteintes cutanées
Le suivi dermatologique est important et régulier dans les premiers mois de la vie : une fois par mois pendant 6 mois puis 2 fois par an jusqu’à 5 ans. Les dermocorticoïdes donnent de bons résultats sur les poussées inflammatoires de stade 1.
Le laser a été utilisé dans le traitement des lésions hyperpigmentées, entraînant une récidive des poussées vésiculaires. Une photo protection est aussi fortement conseillée.
À noter, que la régression spontanée des lésions de la peau est l’évolution habituelle dans l’Incontinentia Pigmenti.
Pour les atteintes au niveau des ongles, les tumeurs péri-unguéales et sous-unguéales l’excision chirurgicale, associé à un curetage de l’os est le traitement habituel. Un traitement au laser CO2 peut aussi être envisagé.
La prise en charge de l’Incontinentia Pigmenti par les centres de la filière FIMARAD ?
Les patients atteints d’IP peuvent être pris en charge au sein :
➔ Du centre de référence des maladies rares de la peau et des muqueuses d’origine génétique (MAGEC) – Nord, à l’hôpital Necker :
Hôpital Necker-Enfants Malades
Service de dermatologie
149 rue de Sèvres
75743 Paris cedex 15
Secrétariat
Premier RDV – Tél. : 01 44 49 46 68
Suivi – Tél. : 01 44 49 43 37
Fax : 01 44 49 44 71
E-mail : dermatologie.rdv@nck.aphp.fr
Les documents de la filière
➔ PNDS Incontinentia Pigmenti
➔ PNDS interactif
➔ Synthèse à destination du médecin traitant
Voir aussi
➔ La page Orphanet sur l’Incontinentia Pigmenti
➔ La page Orphanet sur la dysplasie ectodermique
Au niveau des atteintes dentaires
Le traitement des atteintes dentaires a pour objectif principal de permettre ou de rétablir les fonctions essentielles que sont celles de la mastication, de la phonation et du sourire.
Chez l’enfant, la prise en charge bucco-dentaire permet de traiter la perte des dents temporaires ou définitives, ainsi que les anomalies de structure. La pose de prothèse dentaire pédiatrique est possible dès le plus jeune âge.
Chez l’adulte, une prise en charge parodontale est souvent nécessaire. Le remplacement des dents manquantes est réalisé grâce à des prothèses sur implant dentaire.
Les patients atteints d’Incontinentia Pigmenti peuvent être prises en charge au sein des centres suivants :
➔ Les Centres de Référence ou de Compétence des Maladies Rares Orales et Dentaires (O-Rares), pour leurs soins dentaires.
➔ Les Centres de Référence ou de Compétence des Fentes et Malformations Faciales (MAFACE), pour la prise en charge des malformations faciales.
➔ Les Centres de Référence ou de Compétence des Syndromes de Pierre Robin et Troubles de succion-déglutition congénitaux (SPRATON), en cas de troubles de l’oralité.
Ces centres font partie de la filière maladies rares Tête Cou (malformations rares Tête Cou Dents).
➔ Un programme d’Éducation Thérapeutique du Patient (ETP) destiné aux enfants et adultes atteints de maladies rares orales et dentaires est proposé par le CRMR coordonnateur O-Rares des Hôpitaux Universitaires de Strasbourg.
Les documents de la filière Tête Cou
➔ PNDS Agénésies dentaires : anodontie et oligodontie
➔ Synthèse à destination du médecin traitant
Au niveau des atteintes oculaires
Un examen ophtalmologique auprès d’un ophtalmologue spécialisé doit être réalisé dès que le diagnostic d’IP est posé avec un suivi recommandé tous les mois jusqu’à 3-4 mois, puis trimestriel pendant 1 an, bi-annuel pendant 3 ans. Au-delà, s’il n’y a pas d’anomalies, le pronostic visuel est bon.
Au niveau des atteintes neurologiques
Un examen neurologique complet est nécessaire pour tout patient atteint d’Incontinentia Pigmenti. Souvent les convulsions dans la petite enfance peuvent être un indicateur de problèmes futurs dans le développement psycho-moteur. La survenue dans l’enfance et/ou l’adolescence de crises d’épilepsie est facilement contrôlable par les traitements anti-épileptiques et n’est pas forcément responsable de retards mentaux.
La prise en charge est donc souvent multiple pour les patients atteints d’Incontinentia Pigmenti. L’enquête familiale est aussi vivement recommandée en recherchant des signes typiques et évocateurs : fausses couches à répétition de fœtus porteur du chromosome Y, décès de garçons en période néo-natale, éruptions cutanées précoces…
Chez les femmes, un conseil génétique est indispensable compte-tenu de la gravité potentielle de la maladie.
Les démarches médico-sociales
Comme toutes les maladies rares, l’IP nécessite souvent de nombreuses démarches administratives qui peuvent s’avérer fastidieuses et parfois décourager les meilleures volontés !
FIMARAD a crée une brochure « Focus sur le dossier MDPH » qui regroupe les informations essentielles pour vous aider à bien constituer votre dossier.
La page « Démarches médico-sociales » du site web fimarad.org reprend aussi l’ensemble des items pour vous aider dans vos démarches et formalités (aides et allocations, affection longue durée, éducation et scolarité, emploi et handicap…). N’hésitez pas à la consulter régulièrement !
Les associations de patients
Les associations de la filière
➔ Association Incontinentia Pigmenti
➔ Association Française des Dysplasies Ectodermiques (AFDE)
➔ Association Anna
Les autres associations
➔ Association pour la Reconnaissance de l’Agénésie Dentaire (ARAD)
Sources
Cette page sur l’Incontinentia Pigmenti a été réalisée à partir de la vulgarisation des articles scientifiques suivants :
➔ Incontinentia pigmenti familiale : récurrences tardives et lésions cutanées inhabituelles. Sciences du Vivant [q-bio]. 2011. hal-01731777, Claire Poreaux
➔ Incontinentia Pigmenti, Morice-Picard F. & Léauté-Labrèze Ch
➔ Page sur l’Incontinentia Pigmenti de la FSMR Tête Cou
Crédits visuels
1, 4, 6, 7, 8, 9 : © divanessa@orange.fr – 2 : © ResearchGate – 3 : © Wikipédia – 5 : © futuroprossimo